Do you fear your birthday? Do you worry about the fact that your days are numbered? Do you fear aging because you know death could be nearby?
I do, and many others like me do too. We do this because we have “orphan” diseases and know that our lives are numbered unless a miracle occurs and a cure is found.
 My name is Mika J. Covington; I’m 24 years old and I have cystinosis. Cystinosis is a rare “orphan” disease that causes the amino acid cystine to accumulate in the cells. As the cystine accumulates in the cells, it slowly damages organs including the kidneys, liver, thyroid, eyes, muscles, and brain. Pharmaceutical companies’ control which diseases are to receive attention and funding from the medical and research communities by determining how much financial gain they will make from the disease. Pharmaceuticals created the term “orphan” disease.
My name is Mika J. Covington; I’m 24 years old and I have cystinosis. Cystinosis is a rare “orphan” disease that causes the amino acid cystine to accumulate in the cells. As the cystine accumulates in the cells, it slowly damages organs including the kidneys, liver, thyroid, eyes, muscles, and brain. Pharmaceutical companies’ control which diseases are to receive attention and funding from the medical and research communities by determining how much financial gain they will make from the disease. Pharmaceuticals created the term “orphan” disease.
An “orphan” disease is a disease that has not been “adopted” by the pharmaceutical industry. Critics maintain this because there is little financial incentive for the private sector to make and market new medications to treat or prevent them. There are almost 7,000 “orphan” diseases in the United States that collectively affect nearly 30 million people. An average of about 4,288 people for each disease. Specifically, in the case of cystinosis, only 2,000 people in the world are affected and in the United States there are only 500.
 Cystinosis has three forms, nephropathic (infantile), late-onset (intermediate), and ocular (adult). I have nephropathic (infantile) cystinosis, which is the most common and severe form of the disease. Patients with nephropathic cystinosis appears normal at birth, however before one year of age they have excessive thirst and urination, and failure to thrive. They are smaller than others their same age and often tend to be in the lowest percentile or even off the pediatric growth chart. And they have delays with walking and bearing weight. Late-onset cystinosis, kidney symptoms typically become apparent during adolescent years. With ocular cystinosis, cystine crystals are present in the eyes but kidney function remains normal.
Cystinosis has three forms, nephropathic (infantile), late-onset (intermediate), and ocular (adult). I have nephropathic (infantile) cystinosis, which is the most common and severe form of the disease. Patients with nephropathic cystinosis appears normal at birth, however before one year of age they have excessive thirst and urination, and failure to thrive. They are smaller than others their same age and often tend to be in the lowest percentile or even off the pediatric growth chart. And they have delays with walking and bearing weight. Late-onset cystinosis, kidney symptoms typically become apparent during adolescent years. With ocular cystinosis, cystine crystals are present in the eyes but kidney function remains normal.
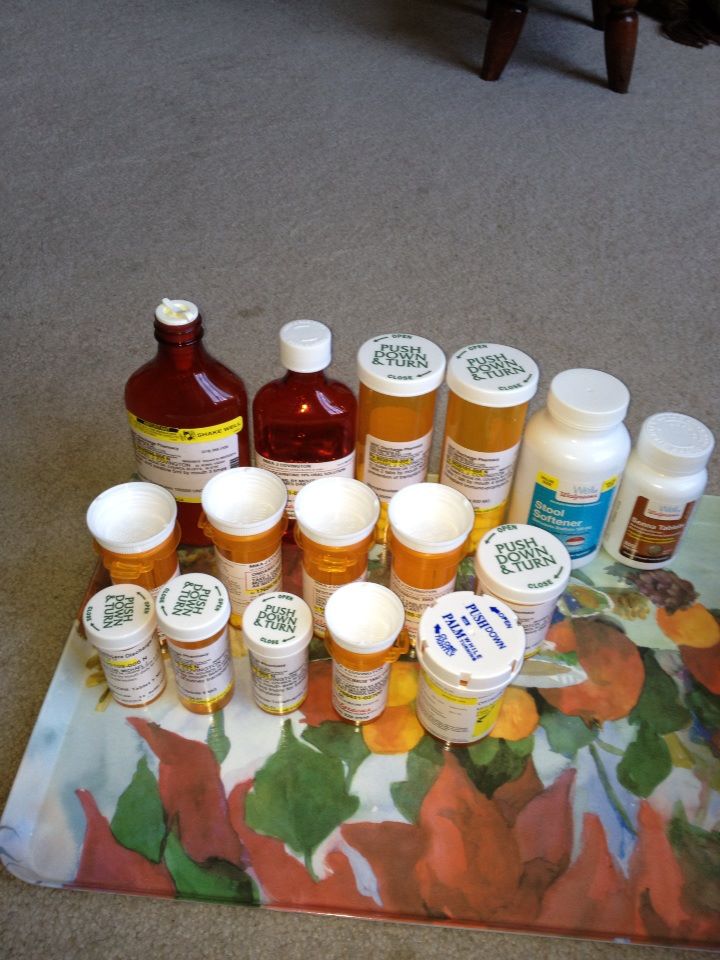 Unfortunately, cystinosis has only one treatment, cysteamine. Cysteamine slows the progression of the disease by removing the cystine from the cells. There are only two forms of the medication, Cystagon and Procysbi. Cystagon was approved by the Food and Drug Administration (FDA) in 1994, and must be taken every six hours every day and has many side effects. Procysbi was approved by the FDA in 2013 for the treatment of only nephropathic cystinosis in adults and children six years and older. Procysbi is a delayed-release form of cysteamine that must be taken every 12 hours every day with many of the similar side effects as Cystagon. However, there is only one treatment for the corneal cystine crystal accumulation, Cystaran. Cystaran was approved by the FDA in 2012. The medication must be put in the eyes, one drop in each eye every hour while awake.
Unfortunately, cystinosis has only one treatment, cysteamine. Cysteamine slows the progression of the disease by removing the cystine from the cells. There are only two forms of the medication, Cystagon and Procysbi. Cystagon was approved by the Food and Drug Administration (FDA) in 1994, and must be taken every six hours every day and has many side effects. Procysbi was approved by the FDA in 2013 for the treatment of only nephropathic cystinosis in adults and children six years and older. Procysbi is a delayed-release form of cysteamine that must be taken every 12 hours every day with many of the similar side effects as Cystagon. However, there is only one treatment for the corneal cystine crystal accumulation, Cystaran. Cystaran was approved by the FDA in 2012. The medication must be put in the eyes, one drop in each eye every hour while awake.
I have been on Cystagon for nearly 22 years of my life. I was on the drug for a part of the clinical research trial before it was approved by the FDA. Cystagon kept me alive, however it made me sick. It caused me to have nausea, vomiting, gastrointestinal issues, headaches, bad breath and body odor. The medication did this because Cystagon dissolves quickly in the stomach causing the harsh chemical make-up to affect the stomach, which has been commonly known to cause stomach ulcers. Taking Cystagon, we don’t get a break because it must be taken every six hours every day. Now, I am on Procysbi.
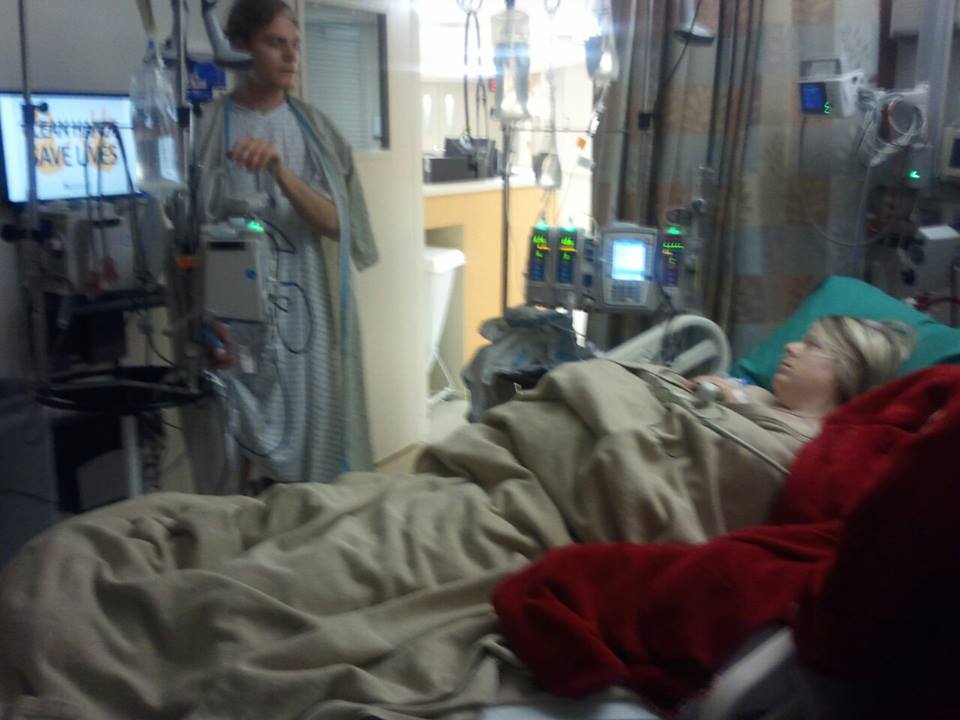
Another major treatment for cystinosis is a kidney transplant, which has become a standard in the treatment of cystinosis because cysteamine only slows the progression of the disease. If not treated by cysteamine, kidney failure occurs at about 12 years old or younger. With cysteamine treatment, the damage of the disease commonly leads to kidney failure by the late teens. Most of the patients must be placed on dialysis to keep them alive before they receive their kidney, which causes more pain, and suffering; sometimes death occurs while waiting for the kidney.
 Nevertheless, being an “orphan” disease, cystinosis does not get the attention or research funding it need to help find better treatments or a cure. All the while, children and young adults are dying from this disease. Compare cystinosis to the Human Immunodeficiency Virus/Acquired Immunodeficiency Syndrome (HIV/AIDS), which affects 1.1 million people in the United States. Persons with HIV/AIDS tend to have a longer life-span than cystinosis patients. HIV/AIDS has more attention and gets more research funding for two reasons. First, it affects more people and thus is more profitable for pharmaceuticals to do so. (I fully recognize that this use to not be true and many people with HIV/AIDS face stigma.) Second, because of the larger number of people with HIV/AIDS, the more people are buying their products. (This is not a good thing; I believe that we should rid the world of HIV/AIDS too.) This is unlike many “orphan” disease, and especially unlike cystinosis.
Nevertheless, being an “orphan” disease, cystinosis does not get the attention or research funding it need to help find better treatments or a cure. All the while, children and young adults are dying from this disease. Compare cystinosis to the Human Immunodeficiency Virus/Acquired Immunodeficiency Syndrome (HIV/AIDS), which affects 1.1 million people in the United States. Persons with HIV/AIDS tend to have a longer life-span than cystinosis patients. HIV/AIDS has more attention and gets more research funding for two reasons. First, it affects more people and thus is more profitable for pharmaceuticals to do so. (I fully recognize that this use to not be true and many people with HIV/AIDS face stigma.) Second, because of the larger number of people with HIV/AIDS, the more people are buying their products. (This is not a good thing; I believe that we should rid the world of HIV/AIDS too.) This is unlike many “orphan” disease, and especially unlike cystinosis.

We must find a cure for all of these “orphan” diseases and certainly for cystinosis. Thus, I have been participating in a long-term clinical research study at the National Institutes of Health (NIH) in Bethesda, Maryland. In the study, I see Dr. William Gahl, one of the leading researchers in the world on cystinosis. Dr. Gahl’s study was one of the first studies of its kind on cystinosis. I currently see Dr. Gahl for the specialized study of the progression of the disease, which I hope will assist in finding a cure. I began seeing Dr. Gahl when I was a baby, shortly after I was diagnosed with cystinosis. I would spend a couple of weeks at a time there. Basically, I grew up there from 1992-1996. In addition, I have participated in several other studies, including at the University of California at San Diego Medical Center, where they studied the neurological and psychological effects of cystinosis.
Today, I am raising funds to help find that cure for my terrible disease. The money raised will go directly to the Cystinosis Research Foundation (CRF). CRF is a non-profit 501c3 organization that was started by the amazing Stack family in 2003. this was after their daughter Natalie Stack made a wish on the eve of her twelfth birthday,
“to have my disease go away forever.”
 CRF today supports bench and clinical research that is focused on developing improved treatments and a cure for cystinosis. CRF has funded every bench and clinical research study that led to Procysbi, allowing cystinosis patients like me to take the drug every 12 hours instead of every 6 hours, which has improved our quality of life. They established the CRF Cystinosis Gene Therapy Consortium, whose mission is to bring stem cell and gene therapy to clinical trial. They also work on effects of cystinosis on neurological function and cognitive development, causes of muscle-wasting and potential therapies.
CRF today supports bench and clinical research that is focused on developing improved treatments and a cure for cystinosis. CRF has funded every bench and clinical research study that led to Procysbi, allowing cystinosis patients like me to take the drug every 12 hours instead of every 6 hours, which has improved our quality of life. They established the CRF Cystinosis Gene Therapy Consortium, whose mission is to bring stem cell and gene therapy to clinical trial. They also work on effects of cystinosis on neurological function and cognitive development, causes of muscle-wasting and potential therapies.
Finding a cure may save my life, as well as others with cystinosis including my sister Mary, or even persons with other diseases. Pharmaceutical companies may not make a profit off the knowledge discovered by studying one “orphan” disease, however often those discoveries leads to advancements in other diseases.
To help find that cure, please make any kind of donation you are able to the Cystinosis Research Foundation (CRF) on my behalf here: Hope Through Research
*All funds donated going directly to research.*


You must be logged in to post a comment.